Research
Our research has been focusing for more than 15 years on omics data sciences for systems biology and precision medicine, with a particular emphasis on metabolomics data sciences.
Data sciences provide powerful approaches and algorithms (signal processing, data mining, machine learning, artificial intelligence) for the processing and analysis of high-dimensional data, such as omics datasets. Metabolomics (untargeted analysis of small molecules involved in biochemical reactions) is of major interest for phenotype characterization and biomarker discovery. High-resolution mass spectrometry (HRMS) is a technology of choice for metabolomics (and also for proteomics), due to its sensitivity and resolution.
We have implemented our new methods in a comprehensive digital ecosystem for metabolomics data sciences, with applications to precision medicine (including neurosciences, liver diseases and food allergy).
Data processing
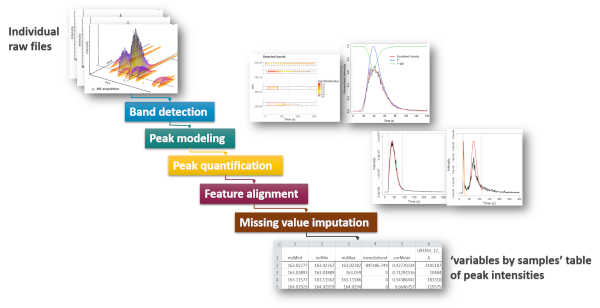
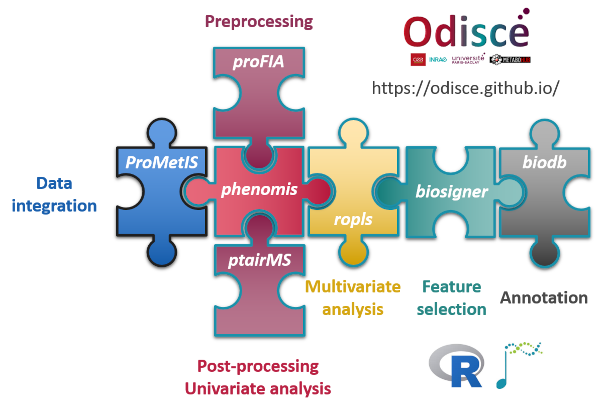
Direct injection methods (such as Flow Injection Analysis or Proton Transfer Reaction) are of particular interest for high-throughput phenotyping. We therefore developed an innovative preprocessing workflow which takes as input the individual raw files and generates the samples by variables table of intensities (peak table). The steps include (i) peak detection and quantification within each file, (ii) peak alignment across samples to generate the peak table, and (iii) missing value imputation. In particular, new methods were required to optimize step (i), including noise estimation, modeling of the injection peak, and precise determination of each analyte peak borders. Application to several real data sets resulted in robust and accurate detection and quantification. proFIA is available as an R/Bioconductor package (Delabrière et al, 2017).
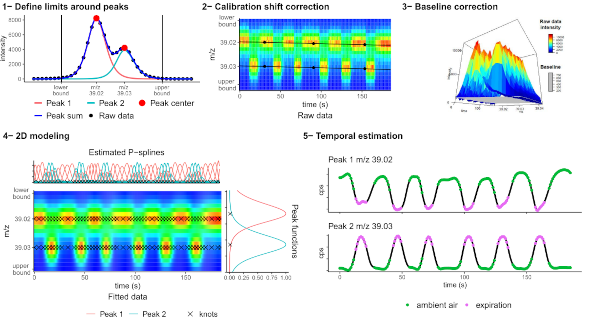
In volatolomics (analysis of volatile organic compounds), PTR-TOF-MS offers unique opportunities for real-time analysis of exhaled air at the patient’s bedside (Grassin Delyle et al., 2021). We therefore developed a comprehensive suite of algorithms for the pre-processing of acquisitions in large cohorts, which includes an innovative two-dimensional peak deconvolution model based on penalized splines signal regression for accurate estimation of the temporal profile, implemented in the ptairMS software (Roquencourt et al, 2022).
Machine learning
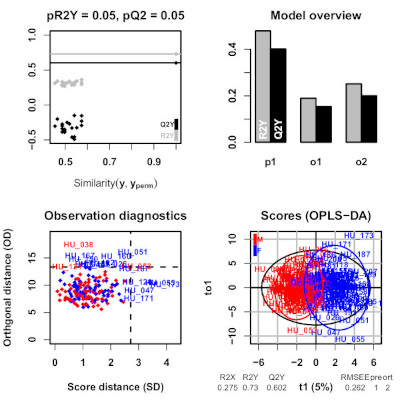
We implemented the Orthogonal Partial Least-Squares (OPLS) approach for regression and classification from Trygg and Wold (2002) as an R package named ropls (Thévenot et al, 2015). OPLS algorithm is a variation of PLS and allows to model separately the orthogonal variation (i.e. non-correlated to the response) and the predictive variation (i.e. correlated to the response), and thus facilitates model interpretation.
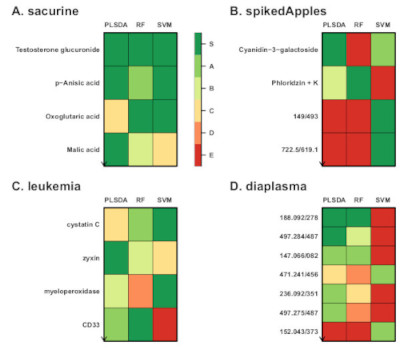
We developed a new methodology for feature selection, of the wrapper type, which assesses the significance of the features for the model performance (biosigner R package). The wrapping of three classifiers (PLS-DA, Random Forest and Support Vector Machine) with this methodology resulted in stable signatures of restricted size, when applied to real metabolomics and transcriptomics datasets (Rinaudo et al, 2016).
Integration of complementary omics data is an opportunity to build more robust predictive models and facilitate the biologicial interpretation. To demonstrate the feasibility and interest of combining proteomics and metabolomics in routine, we have developed, within a consortium of research infrastructures in phenogenomics (PHENOMIN), proteomics (ProFI), metabolomics (MetaboHUB) and bioinformatics (IFB), a data post-processing and integration pipeline that we have applied to the study of murine models. All the data and codes are available in the ProMetIS package (Imbert et al., 2021).

Cheminformatics
The search for similarities within a collection of MS/MS spectra is a powerful approach to facilitate the identification of new metabolites. We developed an innovative de novo strategy for searching for exact fragmentation patterns within such MS/MS spectral libraries. This approach is based on (i) a new representation of spectra as graphs of m/z differences, and (ii) an efficient frequent-subgraph mining algorithm. These new fragmentation patterns capture similarities that are not extracted by existing methods, and facilitate the structural interpretation of molecular network components and the elucidation of unknown spectra. The method is publicly available in the mineMS2 software library (Delabrière et al., 2025).
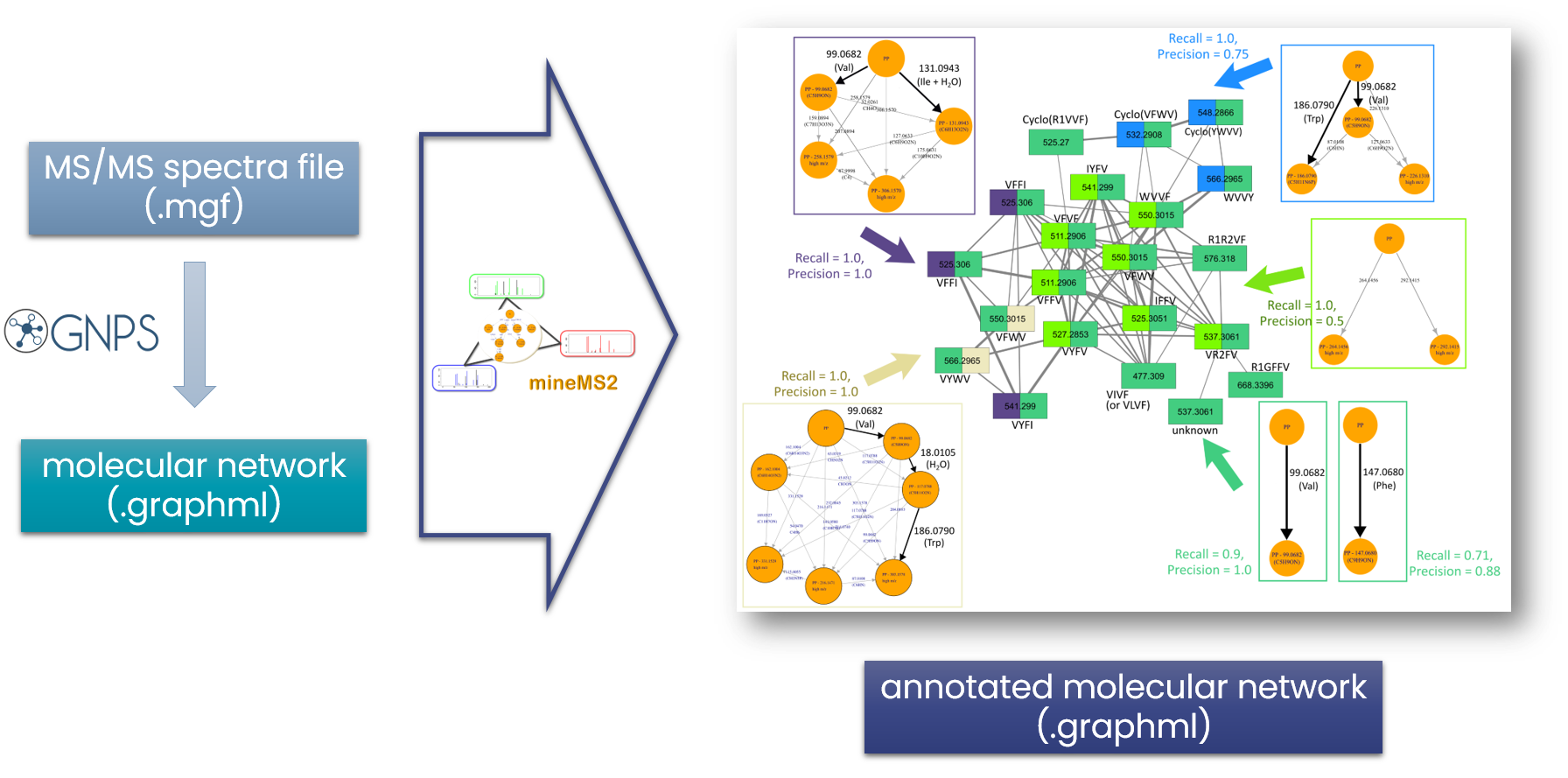
Workflow management
Reference datasets are available in the MetaboLights repository as well as in R packages (plasFIA, ptairData, ropls, biosigner, ProMetIS).
All algorithms are implemented as a comprehensive suite of 6 R packages on the Bioconductor repository.
Many of them are also available as Galaxy modules on the Workflow4Metabolomics online platform,jointly developed and maintained by the French Institute of Bioinformatics and MetaboHUB (Giacomoni et al, 2015; Guitton et al, 2017).